BioFabUSA is committed to assisting its members to ruminate on all potential regulatory pathways and market strategies for their product, and as such would like to highlight unique benefits presented to sponsors who utilize drug master files (DMFs). A DMF is a voluntary submission sent to either the Center for Biologics Evaluation and Research (CBER) or the Center for Drug Evaluation and Research (CDER) that acts as a secure “file cabinet” for the proprietary information stored within different sections in the DMF. Confidential DMF information could include intellectual property (IP), detailed facility descriptions, and even toxicology data for drug substances and products. When filed within a DMF, confidential information may be presented to the FDA without disclosing the material to other product developers and manufacturers who wish to reference that information in their own regulatory submission. It is important to recognize that DMFs can support, but not replace, an application.
The table below shows the different types of DMFs used to designate the subject of the information that the DMF holder wishes to provide.
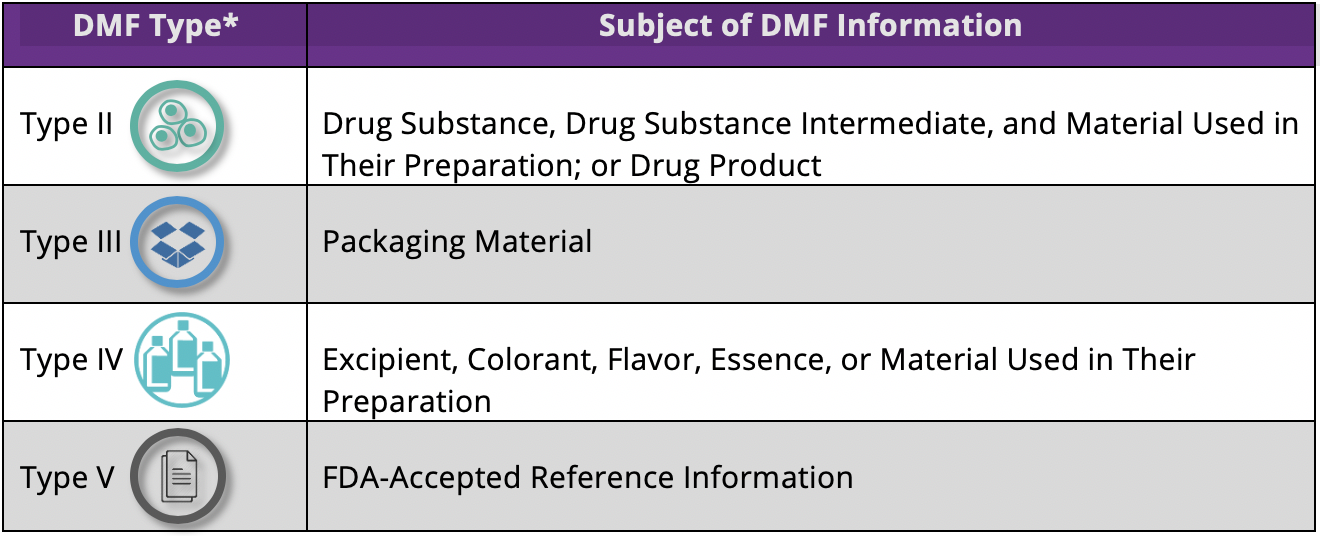
Five types of drug master files were originally used, but Type I DMFs were discontinued in 2000. The numbering of the four DMF types that are currently in use has not changed.
An example case in which the use of a drug master file would be beneficial for a manufacturer is when the manufacturer has a technology, such as a scaffold, that other product developers could take advantage for their own product. Submission of a Type II DMF would protect the scaffold manufacturer’s confidential information from the product developer seeking to use the scaffold, but still permit the FDA to review the referenced information on the scaffold. This exchange of information between the scaffold manufacturer, product developer, and the FDA is governed by a letter of authorization (LOA). The LOA permits the FDA to review the DMF and allows the authorized party to incorporate the information into a regulatory application by reference.
To learn more about the structure, contents, and submission process for drug master files, refer to the newly released FDA draft guidance document, “Drug Master Files”. ARMI|BioFabUSA members are encouraged to submit comments and suggestions regarding this draft guidance document to the Docket prior to December 18, 2019. Comments may be submitted to the Docket via the Federal Register Notice webpage or mailed to the Agency. Because all comments and supporting documents submitted to the docket will be published and made available to the public, you should not include company confidential information in your comment.
The use of a DMF in your regulatory strategy could influence how your applications are reviewed and how your product is used. Therefore, it may be helpful to strategize with a knowledgeable regulatory professional before submitting a DMF to ensure that this is the best path for your product and that you are choosing the correct DMF type. Specific BioFabUSA member questions regarding DMF use, structure, or submission may be addressed in BioFabConsulting meetings with Drs. Richard McFarland and Becky Robinson-Zeigler. BioFabConsulting’s regulatory experts are ready to assist you at any stage of DMF creation, including complete DMF writing and eCTD submission services.



